Getting Started¶
The CellOrganizer project provides tools for
- learning generative models of cell organization directly from images
- storing and retrieving those models in XML files
- synthesizing cell images (or other representations) from one or more models
Model learning captures variation among cells in a collection of images. Images used for model learning and instances synthesized from models can be two- or three-dimensional static images or movies.
CellOrganizer can learn models of
- cell shape
- nuclear shape
- chromatin texture
- vesicular organelle size, shape and position
- microtubule distribution.
These models can be conditional upon each other. For example, for a given synthesized cell instance, organelle position is dependent upon the cell and nuclear shape of that instance. Cell types for which generative models for at least some organelles have been built include human HeLa cells, mouse NIH 3T3 cells, and Arabidopsis protoplasts. Planned projects include mouse T lymphocytes and rat PC12 cells.
Support for CellOrganizer has been provided by grants GM075205, GM090033 and GM103712 from the National Institute of General Medical Sciences, grants MCB1121919 and MCB1121793 from the U.S. National Science Foundation, by a Forschungspreis from the Alexander von Humboldt Foundation, and by the Freiburg Institute for Advanced Studies.
About this document¶
This document covers the minimum materials required for learning model distributions of cellular organization and synthesizing images from those distributions. For more in-depth examples, please refer to the demos and the templates directories contained within the CellOrganizer distribution.
Loading CellOrganizer¶
To start using CellOrganizer, start a Matlab session and change directory to the location of CellOrganizer and run setup.m. In the Matlab terminal, type:
cd( ‘/path/to/cellorganizer’ );
setup;
If you were successful you should either see a message like:
>> setup
Checking for new stable version... Version is up to date.
or see
Tutorials¶
- For detailed documentation of the tutorial give at MMBioS Computational Methods for Spatially Realistic Microphysiological Simulations 2.45 click here
Demos¶
2D/3D Demos¶
For convenience, a series of demos are included with each distribution of CellOrganizer. These demos show
- how synthesize images from existing models,
- how to train new models from raw data, as well as
- other functionality, e.g. exporting examples in multiple formats.
To display information about the available demos contained in the distribution, type in Matlab terminal:
>> demoinfo
This is the documentation associated with each demo:
demo2D00
Synthesize one 2D image from all object models. Results will be one TIFF file, with six slices, one each for cell boundary, nuclear boundary, nucleoli, mitochondria, lysosomes, and endosomes
demo2D01
Trains a 2D generative model of protein location using the 2D HeLa dataset provided
by the Murphy Lab at http://murphylab.web.cmu.edu/data/
demo2D02
Synthesize one 2D image from the LAMP2 model trained in demo2D01
demo2D03
Trains a 2D generative model of protein location using the 2D HeLa dataset provided
by the Murphy Lab at http://murphylab.web.cmu.edu/data/ while making
pretty plots
demo3D00
Synthesizes 1 image using a lysosomal model with sampling mode
set to 'disc' and no convolution.
Results will be three TIFF files, one each for cell boundary,
nuclear boundary, and lysosomes, in folder "synthesizedImages/cell1"
demo3D01
Synthesize one 3D image from all object models,
with sampling mode set to 'disc' and no convolution.
Results will be six TIFF files, one each for
cell boundary, nuclear boundary, nucleoli, mitochondria, lysosomes,
and endosomes, in folder "synthesizedImages/cell1"
demo3D02
Take results from demo3D00 in
folder "../demo3D00/synthesizedImages/cell1"
and generate surface plot
demo3D03
Synthesize one 3D image from all object models,
with sampling mode set to 'sampled' at a density of 75 and no convolution.
Results will be six TIFF files, one each for
cell boundary, nuclear boundary, nucleoli, mitochondria, lysosomes,
and endosomes, in folder "synthesizedImages/cell1"
demo3D04
Synthesize one 3D image from microtubule model and no convolution.
Results will be three TIFF files, one each for
cell boundary, nuclear boundary, and microtubules,
in folder "synthesizedImages/cell1"
demo3D05
Synthesize one 3D image from all object models and a microtubule model,
with sampling mode set to 'sampling' and no convolution.
Results will be seven TIFF files, one each for
cell boundary, nuclear boundary, nucleoli, mitochondria, lysosomes,
endosomes, and microtubules, in folder "synthesizedImages/cell1"
demo3D06
Synthesize one 3D image from all object models and a microtubule model,
with sampling mode set to 'disc' and convolution with point-spread function.
Results will be seven TIFF files, one each for
cell boundary, nuclear boundary, nucleoli, mitochondria, lysosomes,
endosomes, and microtubules, in folder "synthesizedImages/cell1"
demo3D07
Synthesize one 3D image from all object models and a microtubule model,
with sampling mode set to 'sampled' at a density of 25 and
convolution with point-spread function.
Results will be seven TIFF files, one each for
cell boundary, nuclear boundary, nucleoli, mitochondria, lysosomes,
endosomes, and microtubules, in folder "synthesizedImages/cell1"
demo3D08
Synthesize one 3D image from all object models and a microtubule model,
with sampling mode set to 'disc' without convolution.
Results will be an indexed image in a single TIFF file, with one index
each for cell boundary, nuclear boundary, nucleoli, mitochondria, lysosomes,
endosomes, and microtubules, in folder "synthesizedImages/cell1"
demo3D09
Synthesize one 3D image from a lysosome model,
with sampling mode set to 'disc' without convolution.
Results will be three TIFF files, one each for
cell boundary, nuclear boundary, and lysosomes
in folder "synthesizedImages/cell1"
Also produce a mean projection of the cell boundary in
XY, XZ and YZ directions and save it in file 'projection.tif'
demo3D10
Synthesize 1 instance using a lamp2 model with sampling mode
set to 'disc' and no convolution.
Results will be three .obj files, one each for
cell boundary, nuclear boundary, and lamp2,
in folder "synthesizedImages/cell1"
demo3D11
Trains a generative model of the cell framework using the four patterns in the 3D HeLa
dataset from the Murphy Lab
demo3D12
Trains a generative model of the framework using one of the four patterns in the HeLa
dataset
demo3D13
This demo show the usage of syn2blender, a helper method that takes a
folder of synthesized images and exports the images as object files
that can be imported in Blender. This demo uses the images in demo3D03
demo3D14
This demo show the usage of syn2projection, a helper method that makes
projection using a folder of synthesized images
demo3D15
Synthesizes 1 image using a transferrin model for the protein and a diffeomorphic model
for the nuclear and cell shape
Results will be three TIFF files, one each for cell boundary,
nuclear boundary, and protein, in folder "synthesizedImages/cell1"
demo3D16
This method shows how to preprocess raw images to use as input for
CellOrganizer. The main idea behind this demo is to show the user they
can use their own binary images from raw experimental data they can use
to synthesize protein patterns. The current demo assumes the resolution
of the images is the same as the images that were used to train the
protein model
This method shows how to input an image to CellOrganizer.
The main idea behind this demo is to show the user they
can use their own binary images from raw experimental data. They can use
them to synthesize protein patterns. The current demo assumes the resolution
of the images is the same as the images that were used to train the
protein model. This demo uses the framework synthesized from demo3D15. In
this case, the resolution at which the diffeomorphic and vesicle model were
trained on are different. This demo also shows how to handle that situation
in CellOrganizer
demo3D18
Trains a generative model of the framework using the holefinding
functionality
demo3D19
This method shows the use of slml2report for creating comparisons between
parameters of CellOrganzier models.
demo3D20
Trains a generative model of the framework using one diffeomorphic model
demo3D21
Trains a generative model of the framework using the holefinding
functionality. The same demo as demo3D18 but with no scaling of the
images.
demo3D22
Synthesizes a protein pattern instance for each of the synthetic images
from demo3DDiffeoSynth
demo3DMultiresSynth
Synthesize multiple 3D images from a lysosome model,
at different resolutions
demo3DObjectAvoidance
Synthesizes 1 image using a lysosomal model with sampling mode
set to 'disc', no convolution using the object avoidance methods
Results will be three TIFF files, one each for cell boundary,
nuclear boundary, and lysosomes, in folder "synthesizedImages/cell1"
demo3DPrimitives
Synthesizes 1 image using a lysosomal model with sampling mode
set to 'disc', no convolution and output.SBML set to true
Results will be three TIFF files, one each for cell boundary,
nuclear boundary, and lysosomes, in folder "synthesizedImages/cell1"
Additionally, in the folder "synthesizedImages/" will be a
SBML-Spatial(v0.82a) formatted .xml file containing constructed solid
geometry(CSG) primitives for lysosomes and parametric objects for the
cell and nuclear shapes.
These files can then be read into VCell using the built in importer or
CellBlender using the helper function provided in this distribution.
Contents of demo3DDiffeoSynth:
demo3DDiffeoPick - function [ output_args ] = demo3DDiffeoSynth( input_args )
demo3DDiffeoSynth - This demo illustrates different ways to sample from points in a
demo3DDiffeoSynth_gmm - function [ output_args ] = demo3DDiffeoSynth( input_args )
demo3DDiffeoSynth_grid - function [ output_args ] = demo3DDiffeoSynth( input_args )
demo3DDiffeoSynth_grid_pick - function [ output_args ] = demo3DDiffeoSynth( input_args )
demo3DDiffeoSynth_uniform - function [ output_args ] = demo3DDiffeoSynth( input_args )
demo3DDiffeoSynth is both a directory and a function.
This demo illustrates different ways to sample from points in a
diffeomorphic model.
Contents of demo3DDiffeomorphicWindowSize:
demo3DDiffeomorphicWindowSize - This demo exists to illustrate how padding size and window size effect the
demo3DDiffeomorphicWindowSize is both a directory and a function.
This demo exists to illustrate how padding size and window size effect the
performance of diffeomorphic metric. Currently incomplete GRJ 4/16/14
todo: output results
This program is free software; you can redistribute it and/or modify
it under the terms of the GNU General Public License as published
by the Free Software Foundation; either version 2 of the License,
or (at your option) any later version.
This program is distributed in the hope that it will be useful, but
WITHOUT ANY WARRANTY; without even the implied warranty of
MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU
General Public License for more details.
You should have received a copy of the GNU General Public License
along with this program; if not, write to the Free Software
Foundation, Inc., 51 Franklin Street, Fifth Floor, Boston, MA
02.40-1301, USA.
For additional information visit http://murphylab.web.cmu.edu or
send email to murphy@cmu.edu
Contents of demo3DDynamic:
demo3DDynamic -
demo3DDynamic is both a directory and a function.
demo3DDynamic
Learns a random walk from time series images as in figure 6 and 7 of
Johnson 2.45
Contents of demo3DSBML:
demo3DSBML - This demo converts a sample SBML file to an SBML-spatial instance using
makeCSGOnly - This function makes the SBML-spatial standard 'CSGOnly.xml' file.
makeMeshOnly - This function makes the SBML-spatial standard 'MeshOnly.xml' file.
makeMixedOnly - This function makes the SBML-spatial standard 'MixedOnly.xml' file.
demo3DSBML is both a directory and a function.
This demo converts a sample SBML file to an SBML-spatial instance using
the "matchSBML" function. This function takes an SBML file, matches the
compartments in the file with available models and synthesizes the
appropriate instances
This demo is to demonstrate the function that reads an image and creates a
3D mesh using the SBML-spatial paradigm.
Image Synthesis Walkthrough¶
demo3D01¶
If you are interested in learning how to use slml2img to synthesize an image from multiple models, then you should explore demo3D01 first.
To run this demo, type:
cd( ‘/path/to/cellorganizer’ );
setup;
demo3D01();
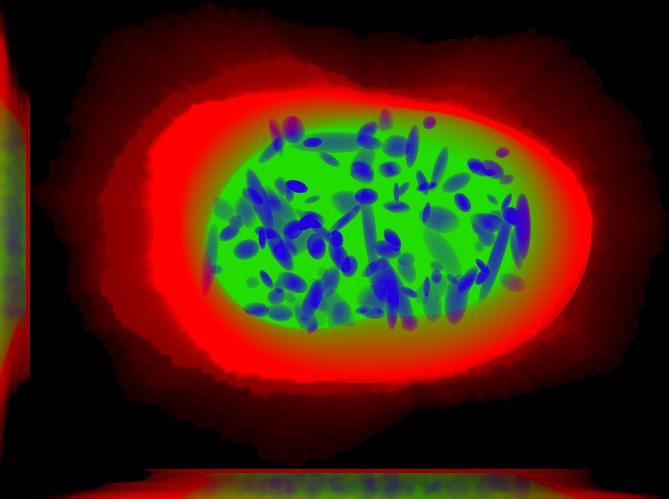
This is a sum projection that includes the nucleus, the cell boundary and nucleoli. This image was generated using img2projection.
All demos are seeded, meaning that when you run them, you should get the same results shown here and included in the distribution. This demo synthesizes one image saved to disk as multiple tiff files. Each tiff file correspond to one channel. Because this demo uses four models we should expect to find six tiff files. One for the nuclear channel, one for the cell boundary and then four files; one for each protein pattern included in each of the files:
function answer = demo3D01( param )
% demo3D01
%
% Synthesize one 3D image from all object models,
% with sampling mode set to 'disc' and no convolution.
% Results will be six TIFF files, one each for
% cell boundary, nuclear boundary, nucleoli, mitochondria, lysosomes,
% and endosomes, in folder "synthesizedImages/cell1"
curr_path = which('demo3D01.m');
curr_path = curr_path(1:end-10);
cd(curr_path);
outputDirectory = pwd;
if nargin == 0
param = [];
end
param = ml_initparam( param, ...
struct( 'numberOfSynthesizedImages', 1 ) );
param = ml_initparam( param, ...
struct( 'seed', 3 ) );
param.targetDirectory = outputDirectory;
param.prefix = 'synthesizedImages';
param.compression = 'lzw';
param.microscope = 'none';
param.sampling.method = 'disc';
param.verbose = true;
param.debug = false;
try
state = rng( param.seed );
catch
state = RandStream.create('mt19937ar','seed', param.seed );
RandStream.setDefaultStream(state);
end
answer = slml2img( {'../../../models/3D/lamp2.mat', ...
'../../../models/3D/mit.mat', ...
'../../../models/3D/nuc.mat', ...
'../../../models/3D/tfr.mat'}, param );
This demo allows you to pass in two parameters
- param.numberOfSynthesizedImages. Default value is 1. Can be any integer.
- param.seed. Default value is 3. Use any integer to generate a new stream of pseudo-random numbers.
For example, you can run:
cd( ‘/path/to/cellorganizer’ );
setup;
param.numberOfSynthesizedImages = 25;
demo3D01();
to synthesize 25 images with the same seed, or:
cd( ‘/path/to/cellorganizer’ );
setup;
param.seed = 5;
demo3D01();
to synthesize one image that looks different from the one included with the distribution, or:
cd( ‘/path/to/cellorganizer’ );
setup;
param.numberOfSynthesizedImages = 25;
param.seed = 5;
demo3D01();
to generate multiple images with a different seed.
API¶
These is the documentation attached to each of the main methods
img2slml¶
Method header:
function answer = img2slml( varargin )
% IMG2SLML Trains a generative model of protein subcellular location from a
% collection of microscope images and saves the model as an SLML instance. An SLML model
% consists of four components, a (optional) documentation component, a nuclear pattern model,
% a cell pattern model and a protein pattern model. The input argument param holds the valid
% options for these components.
%
% Arguments Description
% --------- ------------
% dimensionality 2D/3D
% dnaImagesDirectoryPath DNA images collection directory
% cellImagesDirectoryPath Cell images collection directory
% proteinImagesDirectoryPath Protein images collection directory
% param Parameter structure
%
% The shape of param is described
%
% List Of Parameters Descriptions
% ------------------ ------------
% documentation (optional)
% ------------------------
% This is an optional structure with multiple elements that holds documentation about this model.
%
% documentation.<name> Holds the value of variable <name>. This is meant to be meta information. Default is empty.
%
% generic model options
% ---------------------
% model.name (optional) Holds the name of the model. Default is empty.
% model.id (optional) Holds the id of the model. Default is empty.
% model.filename Holds the output filename.
% model.resolution Model resolution (in microns per pixel). This
% the resolution of the dataset used to train the model
% model.downsampling Downsampling vector used during preprocessing. Default value is
% [5 5 1]. Final model resolution will be resolution * downsampling
% vector and will be saved in the model as well
%
% nuclear shape model options
% ---------------------------
% nucleus.type Holds the nuclear model type. Default is
% "medial axis" for 2D and "cylindrical_surface" for 3D
% nucleus.name (optional) Holds the name of the nuclear model. Default is empty.
% nucleus.id (optional) Holds the id of the nuclear model. Default is empty.
%
% cell shape model options
% ------------------------
% cell.type Holds the cell model type. Default is "ratio".
% cell.name (optional) Holds the name of the cell model. Default is empty.
% cell.id (optional) Holds the id the cell model. Default is empty.
%
% protein shape model options
% ---------------------------
% protein.type (optional) Holds the protein model type. The default is "vesicle".
% protein.name (optional) Holds the name of the protein model. The default is empty.
% protein.id (optional) Holds the id of the protein model. The default is empty.
% protein.class Holds the protein class, e.g. lysosome, endosome.
% protein.cytonuclearflag (optional) Determines whether the protein pattern will be generated in
% the cytosolic space ('cyto'), nuclear space ('nuc') or everywhere ('all').
% Default is cyto.
%
% other options
% -------------
% verbose (optional) Displays messages to screen. Default is true.
% debug (optional) Reports errors and warnings. Default is false.
% train.flag (optional) Selects what model is going to be trained ('nuclear',
% 'framework', or 'all'). Default is 'all'.
slml2img¶
Method header:
function answer = slml2img( varargin )
% SLML2IMG Synthesizes an instance from a collection
% of SLML Level 1.0 Version 1.* models.
% instances may be saved in the following forms:
% Tiff stacks = a 3D tiff image stack for each pattern generated using the input models
% indexed images = a single 3D tiff image stack where each pattern is represented by a number 1-n
% object mesh = a .obj mesh file for each pattern generated using the input models (blenderfile option)
% SBML-Spatial file = a systems biology markup language(SBML) formatted xml file utilizing the spatial extension in level 3 version 1
%
%
% COMMENT: When the method is not deployed the shape of the input arguments is
% slml2img( models, param )
%
% List Of Input Arguments Descriptions
% ----------------------- ------------
% models A cell array of filenames
% param A structure holding the function options
%
% The shape of param is described
%
% List Of Parameters Descriptions
% ------------------ ------------
% targetDirectory (optional) Directory where the images are going to be saved. Default is current directory.
% prefix (optional) Filename prefix for the synthesized images. Default is 'demo'
% numberOfSynthesizedImages (optional) Number of synthesized images. Default is 1.
% compression (optional) Compression of tiff, i.e. 'none', 'lzw' and 'packbits'
% debug (optional) Keeps temporary results and catches errors with full reports
% display (optional) Display flag for figures
% verbose (optional) Print the intermediate steps to screen. Default is true.
% microscope (optional) Microscope model from which we select a point spread function. Default is 'none'
% synthesis (optional) Synthesis parameter that allows to
% synthesize 'nucleus', 'framework' or 'all'. Default is 'all'
% protein.cytonuclearflag (optional) Defines the allowable region for protein placement.
% The default is the cytonuclearflag included in the model.
% sampling.method (optional) Can be 'disc' or 'sampled'. Default is trimmed
% savePDF (optional) Saves the probability density function for a given pattern during 2D synthesis. Default is False.
% spherical_cell (optional) Boolean flag that indicates whether a cell is spherical. Default is False.
% synthesis.diffeomorphic.maximum_iterations (optional) Integer defining the maximum number of iterations during diffeo inference. Default is 100.
% randomwalk (optional) Boolean flag of whether to perform a shape space walk. Default is False.
% framefolder (optional) The folder in which to look for completed frames and save finished frames from the diffeomorphic synthesis.
% The default is './frames/'.
% walksteps (optional) The integer number of steps to walk during a shape space walk. Default is 1.
% walk_type (optional) Type of random walk to perform. Default is 'willmore'.
% overlapsubsize (optional) Defines the downsampling fraction to perform during object overlap avoidance. Default is 0.3.
% overlapthresh (optional) Defines the amount of overlap that is allowed between objects. Default is 1.
% rendAtStd (optional) Defines the number of standard deviations to render gaussian objects at. Default is 2.
% sampling.method.density (optional) An integer. Default is empty.
% protein.cytonuclearflag (optional) Can 'cyto', 'nucleus' or 'all'. Default is all.
% resolution.cell (optional) The resolution of the cell and nucleus that are being passed in
% resolution.objects (optional) The resolution of the object model being synthesized
% instance.cell (optional) A binary cell image to be filled with objects. Default is empty.
% instance.nucleus (optional) A binary nuclear image to be filled with objects. Default is empty.
% image_size (optional) The image size. Default is [1024 1024] for both 2D and 3D in x and y
% spherical_cell (optional) Indicates whether the cell is spherical or not. Default is FALSE
%
% output.tifimages (optional) boolean flag specifying whether to write out tif images
% output.indexedimage (optional) boolean flag specifying whether to write out indexed image
% output.blenderfile (optional) boolean flag specifying whether to write out (.obj) files for use in blender
% output.blender.downsample (optional) downsampling fraction for the creation of object files (1 means no downsampling, 1/5 means 1/5 the size)
% output.SBML (optional) boolean flag specifying whether to write out (.xml) files with SBML-Spatial representations of geometries